本品为伞形科植物积雪草 Centella asiatica(L. )Urb. 的全草经加工制成的总苷。

【性状】本品为淡黄色至淡棕黄色粉末;无臭,味苦、稍具引湿性。
本品在水、乙醇中易溶,在三氯甲烷、乙醚中不溶。
【鉴别】(1)取本品约2mg,置试管中,加醋酐 1ml,摇匀,沿试管壁缓缓加入硫酸1ml,在两液接界处呈紫红色环。
(2)取本品粉末,加乙醇制成每1ml含10mg的溶液,作为供试品溶液。另取羟基积雪草苷对照品、积雪草苷对照品, 分别加乙醇制成每1ml各含10mg的溶液,作为对照品溶液。照薄层色谱法(通则0502)试验,吸取上述三种溶液各5μl,分别点于同一硅胶G薄层板上,以正丁醇-乙酸乙酯-水(4∶1∶5)的 上层溶液为展开剂,展开,取出,晾干,喷以醋酐-硫酸-无水乙醇(1∶1∶10)溶液,在105℃加热至斑点显色清晰。供试品色谱中,在与对照品色谱相应的位置上,显相同的两个蓝褐色斑点。
【检查】干燥失重 取本品,在105℃干燥至恒重,减失重量不得过10.0%(通则0831)。
重金属及有害元素 照铅、镉、砷、汞、铜测定法(通则 2321)测定,铅不得过5mg/kg;镉不得过0.3mg/kg;砷不得 过2mg/kg;汞不得过0.2mg/kg;铜不得过20mg/kg。
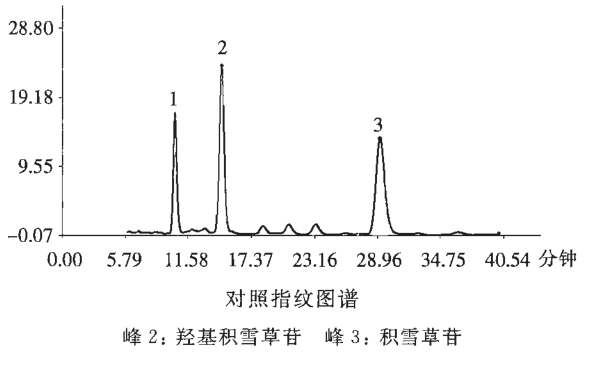
【指纹图谱】照高效液相色谱法(通则0512)测定。
色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂(柱长为25cm,内径为4.6mm,粒径为5μm);以乙腈-2mmol/L倍他环糊精溶液(24∶76)为流动相;检测波长为205nm。理论板数按积雪草苷峰计算应不低于4000。
参照物溶液的制备 取羟基积雪草苷对照品、积雪草苷对照品,精密称定,加甲醇制成每1ml各含0.2mg的溶液, 即得。
供试品溶液的制备 同〔含量测定〕项下的供试品溶液制备。
测定法 分别精密吸取参照物溶液和供试品溶液各10μ1,注入液相色谱仪,测定,记录色谱图,即得。
按中药色谱指纹图谱相似度评价系统,供试品指纹图谱与对照指纹图谱经相似度计算,相似度不得低于0.90。
积分参数 斜率灵敏度为1,峰宽为0.1,最小峰面积为20,最小峰高为0. 5。
【含量测定】照高效液相色谱法(通则0512)测定。
色谱条件与系统适用性试验 以十八烷基硅烷键合硅胶为填充剂;以乙腈-2mmol/L倍他环糊精溶液(24∶76)为流动相;检测波长为205nm。理论板数按积雪草苷峰计算应不低于4000。
对照品溶液的制备 取羟基积雪草苷对照品和积雪草苷对照品适量,精密称定,加甲醇制成每1ml各含0.2mg的溶 液,即得。
供试品溶液的制备 取本品约50mg,精密称定,置50ml量瓶中,加甲醇使溶解,并稀释至刻度,摇匀,滤过,取续滤液,即得。
测定法 分别精密吸取对照品溶液与供试品溶液各 10μ1,注入液相色谱仪,测定,即得。
本品按干燥品计算,含总苷以羟基积雪草苷(C48 H78O20) 和积雪草苷(C48 H78O19)的总量计,不得少于55.0%。
【贮藏】密封。
【制剂】积雪苷片
在线查询结果来源于2020年版中国药典,仅供参考。