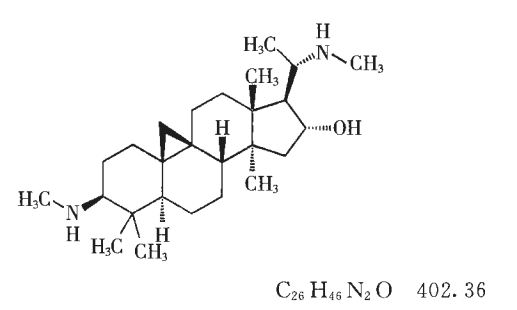
本品为菊科植物短葶飞蓬 Erigeron breviscapus ( Vant.)Hand. -Mazz. 中提取分离所得。按干燥品计算,含野黄苓苷(C21H18O12)不得低于83.5 % (供口服用)或91.0%(供注射用)。

【制法】取灯盏细辛,粉碎成粗粉,加75%乙醇(6倍、4倍、4倍)加热回流提取三次,每次2小时,合并提取液,滤过,滤液浓缩至无醇味,加等体积水搅匀,静置过夜,滤过,滤液通过大孔吸附树脂(聚苯乙烯型)柱,用水洗脱,收集洗脱液,浓缩,沉淀,滤过,沉淀用10%硫酸溶液调pH值至2.0~2.5,静置过夜,滤过,沉淀用乙醇洗涤,再用水洗至中性,干燥,干燥品用乙醇精制,重结晶,结晶用乙醇、丙酮洗涤,干燥,粉碎,混合,即得。或取灯盏细辛粉碎成粗粉,加入2~6倍量75%乙醇,加热回流提取三次,每次3小时,滤过,合并滤液,浓缩至相对密度为1.2(80℃)的清膏,加水适量,搅匀,加热至80℃, 用5%氢氧化钠溶液调节pH值至8,搅拌使溶解,静置24小时,滤过,滤液用10%硫酸溶液调节pH值至1~3,搅拌,静置48小时,抽滤,沉淀用水洗至中性,或先用3~4倍量乙醇洗2~3次,再用水洗涤至中性。加入20倍量85%~95%乙醇及1%量的活性炭,或加入适量甲醇溶解后,加0.1%量的活性炭,加热回流1小时,滤过,滤液浓缩至原体积的60%~80%,静置使析出结晶,滤过,将所得结晶用45%乙醇洗涤5次,于50~80℃减压真空干燥。取结晶物,加水适量,用30% 精氨酸溶液或10%碳酸氢钠溶液调节pH值至7.0~7.5,加热使溶解,离心,取上清液,滤过,滤液通过大孔吸附树脂(聚苯乙烯型)柱,用水洗脱,收集洗脱液,滤过;或用5%盐酸调节pH值至1~3,静置,滤过,沉淀用水洗至中性,取沉淀,加入适量的水搅匀,加热,用20%~30%磷酸氢二钠溶液调节pH值至6.5-7.0,煮沸,冷却至35~55°C ;减压浓缩,加入8~10倍量的丙酮,搅匀,静置,抽滤,用丙酮洗涤沉淀。取沉淀, 加入适量50%~70%丙酮溶液使成混悬液,用10%盐酸溶液调节pH值至1~2,静置,抽滤。取沉淀,用注射用水洗至中性,再用90%乙醇洗涤,烘干,即得。
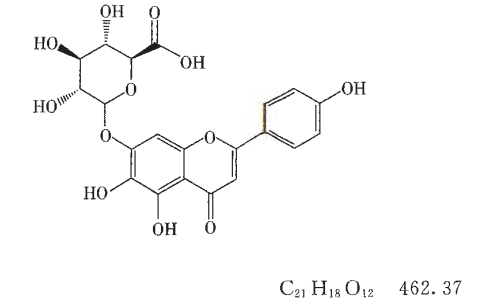
本品在吡啶、稀碱溶液中溶解,在甲醇中微溶,在热水、乙醇、乙酸乙酯中略溶,在水、乙醚、三氯甲烷、苯、丙酮等有机溶剂中几乎不溶。无明显熔点。在284nm±2nm和 335nm±2nm波长处有最大吸收。
【鉴别】照〔含量测定〕项下的方法试验,供试品色谱图中,应呈现与野黄苓苷对照品色谱峰保留时间相同的色谱峰。
【检查】溶液的颜色 取本品,加1%碳酸氢钠溶液溶解并稀释成每1ml含0.02mg的溶液,在5分钟内依法检查, 应澄清,与黄绿色6号标准比色液(通则0901第一法)比较, 不得更深(供注射用)。
干燥失重 取本品约0.5g,置五氧化二磷干燥器中,减 压干燥至恒重,减失重量不得过2.0%(通则0831)。
炽灼残渣 不得过0.5%;供注射用不得过0.2%(通则 0841)。
有关物质 取本品,加1%碳酸氢钠溶液溶解并稀释成每1ml含0.02mg的溶液,除“树脂”外,依法(通则2400)检查,应符合规定(供注射用)。
树脂 取本品,加1%碳酸氢钠溶液溶解并稀释成每1ml 含0.02mg的溶液,取溶液5ml,加三氯甲烷10ml振摇提取, 充分放置,分取三氯甲烷液,置水浴上蒸干,残渣加冰醋酸 2ml使溶解,置具塞试管中,加水3ml,混匀,放置30分钟,不得出现沉淀(供注射用)。
相关物质 照高效液相色谱法(通则0512)测定(供注 射用)。
检查法 取本品适量(相当于野黄苓苷20mg),置50ml 量瓶中,加甲醇适量,超声处理(功率300W,频率50kHz)45分钟,放至室温,加甲醇稀释至刻度,摇匀,作为供试品溶液。精密量取供试品溶液1ml,置100ml量瓶中,加甲醇稀释至刻度,摇匀,作为对照溶液。照〔含量测定〕项下的色谱条件,取对照溶液5μl,注入液相色谱仪,调节检测灵敏度,使主成分色谱峰的峰高为满量程的10%,再精密量取供试品溶液与对照溶液各5μl,分别注入液相色谱仪,记录色谱图至主成分峰保留时间的2.5倍。供试品溶液色谱中,其他成分峰面积的和不得大于对照溶液主峰峰面积的2倍。
丙酮残留物 照残留溶剂测定法(通则0861第二法)测 定(供注射用)。
色谱条件与系统适用性试验 以聚乙二醇为固定相,釆用弹性石英毛细管柱(柱长为30m,内径为0.32mm,膜厚度为0.5μm);柱温为程序升温:初始温度为60℃,维持16分钟,以每分钟20℃升温至200℃,维持2分钟;检测器温度 300℃;进样口温度240℃;载气为氮气,流速为每分钟1.0ml。 顶空进样,顶空瓶平衡温度为90℃,平衡时间为30分钟。理论板数以丙酮峰计算应不低于10000。
对照品溶液的制备 取丙酮对照品适量,精密称定,加 0.5%的碳酸钠溶液制成每1ml含100μg的溶液,作为对照品溶液。精密量取5ml,置20ml顶空瓶中,密封瓶口,即得。
供试品溶液的制备 取本品约0.1g,精密称定,置20ml 顶空瓶中,精密加入0.5%的碳酸钠溶液5ml,密封瓶口,摇匀,即得。
测定法 分别精密量取对照品和供试品溶液顶空瓶气体 1ml,注入气相色谱仪,记录色谱图,按外标法以峰面积计算, 即得。
本品含丙酮不得过0.5%。
大孔吸附树脂有机残留物 正己烷、苯、甲苯、对二甲苯、邻二甲苯、苯乙烯和1,2-二乙基苯 照残留溶剂测定法(通则 0861第二法)测定(供注射用)。
色谱条件与系统适用性试验 以聚乙二醇为固定相,采用弹性石英毛细管柱(柱长为30m,内径为0.32mm,膜厚度 为0.5μm);柱温为程序升温:初始温度为60℃,维持16分钟,以每分钟20℃升温至200℃,维持2分钟;检测器温度 300℃ ,进样口温度240℃;载气为氮气,流速为每分钟2.5ml。顶空进样,顶空瓶平衡温度为80℃,平衡时间为30分钟。理 论板数以邻二甲苯峰计算应不低于10000,各待测峰之间的分离度应符合规定。
对照品溶液的制备 取正己烷、苯、甲苯、对二甲苯、邻二甲苯、苯乙烯和1,2-二乙基苯对照品适量,精密称定,加二甲亚砜制成每1ml中分别含20μg、2μg、20μg、20μg、20μg、 20μg、20μg的溶液,作为对照品储备液。精密量取上述贮备液5ml,置50ml量瓶中,加入2%碳酸钠的25%二甲亚砜溶液稀释至刻度,摇匀,精密量取2ml,置20ml顶空瓶中,密封瓶口,即得。
供试品溶液的制备 取本品约0.2g,精密称定,置20ml 顶空瓶中,精密加入2%碳酸钠的25%二甲亚砜溶液2ml,密封瓶口,摇匀,即得。
测定法 分别精密量取对照品溶液和供试品溶液顶空瓶气体1ml,注入气相色谱仪,记录色谱图,按外标法以峰面积计算,即得。
本品含苯不得过0.0002%,含正己烷、甲苯、对二甲苯、 邻二甲苯、苯乙烯和1,2-二乙基苯均不得过0.002%。
重金属及有害元素 照铅、镉、砷、汞、铜测定法(通则 2321)测定,铅不得过5mg/kg;镉不得过0. 3mg/kg;砷不得 过2mg/kg;汞不得过0. 2mg/kg(供注射用)。
热原 取本品,按100mg加1%碳酸氢钠溶液2.3ml的比例加入1%碳酸氢钠无热原溶液,在50℃水浴振摇使溶解,再加氯化钠注射液制成每1ml含2.5mg的溶液,依法(通则1142)检查, 剂量按家兔体重每1kg注射1ml,应符合规定(供注射用)。
过敏反应 取本品,按100mg加1%碳酸氢钠溶液2.3ml的比例加入1%碳酸氢钠无菌溶液,在50℃水浴振摇使溶解,再加氯化钠注射液制成每1ml中含3mg的溶液,依法(通则1147)检查,应符合规定(供注射用)。
降压物质 取本品,按100mg加1%碳酸氢钠溶液2.3ml 的比例加入1%碳酸氢钠无菌溶液,在50℃水浴振摇使溶解,再加氯化钠注射液制成每1ml含10mg的溶液,依法(通则1145)检查,剂量按每1kg注射0.2ml,应符合规定(供注射用)。
异常毒性 取本品,按100mg加1%碳酸氢钠溶液2.3ml的比例,加1%碳酸氢钠无菌溶液,在50℃水浴振摇使溶解,再加氯化钠注射液制成每1ml含12mg的溶液,依法(通则1141)检查,按静脉注射法给药,应符合规定(供注射用)。
溶血与凝聚 2%红细胞混悬液的制备 取家兔心脏血,置有玻璃珠的容器内,振摇数分钟,除去纤维蛋白原使成脱纤血。加入0.9%氯化钠溶液约10倍量,摇匀,每分钟1000~1500转离心15分钟,倾去上清液,沉淀的红细胞再用0.9%氯化钠溶液按上述方法洗涤3~4次,至上清液不显红色,将所得红细胞用0.9%氯化钠溶液制成2%的混悬液。
溶液的制备 取本品,按每25mg加10%精氨酸溶液 0.1ml溶解,加氯化钠注射液稀释制成每1ml含1mg的溶液。
试验方法 取洁净试管5支,1、2、5号管中各加供试品溶液2.5ml,第3管加0.9%氯化钠溶液2.5ml作为阴性对照管,第4管加蒸馏水2.5ml作为阳性对照管,然后1~4号管分别加2%红细胞混悬液2.5ml,第5管加0.9%氯化钠溶液2.5ml作为供试品对照,摇匀,立即置恒温箱内,保持37℃ 士0. 5℃,在3小时内不得有溶血现象和凝聚现象(供注射用)。

【含量测定】照高效液相色谱法(通则0512)测定。
色谱条件与系统性适用性试验 以十八烷基硅烷键合硅胶为填充剂;以甲醇-0.1%磷酸溶液(40∶60)为流动相;流速为每分钟1.0ml;柱温40℃ ;检测波长335nm。理论板数按野黄苓苷峰计算应不低于5000。
对照品溶液的制备 取野黄苓苷对照品10mg,精密称定,置100ml量瓶中,加甲醇70ml,超声处理(功率300W,频 率50kHz)45分钟,取出,放置室温,加甲醇稀释至刻度,摇匀,即得。
供试品溶液的制备 取本品10mg,精密称定,置100ml 量瓶中,加甲醇70ml,超声处理(功率300W,频率50kHz)45分钟,取出,放置室温,加甲醇稀释至刻度,摇匀,滤过,取续滤 液,即得。
测定法 分别精密吸取对照品溶液和供试品溶液各5μl, 注入液相色谱仪,测定,即得。
【贮藏】遮光,密闭。
【制剂】口服制剂 注射剂
在线查询结果来源于2020年版中国药典,仅供参考。